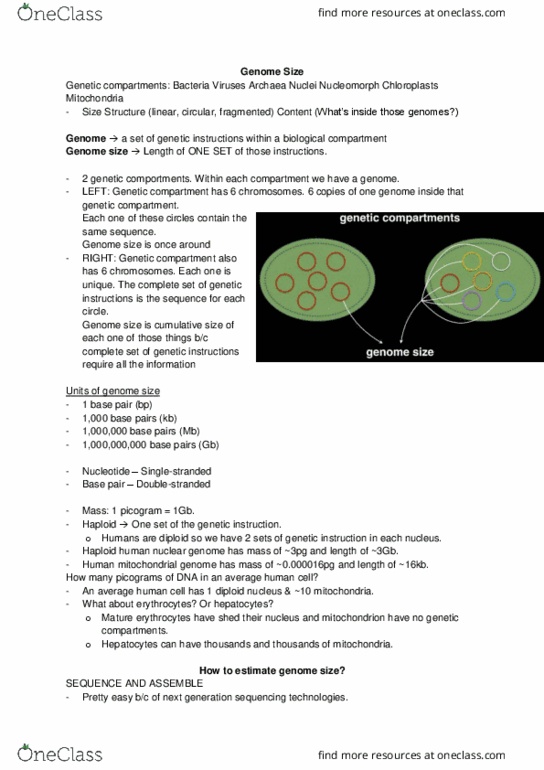
BINF 511 Lecture Notes - Lecture 11: De Bruijn Graph, Sequence Assembly, Contig

25 Jun 2018
School
Department
Course
Professor

Lecture 11: Genome assembly; Extra lab
April 4, 2018
Lecture portion: Introduction to genome assembly (lecturer: Maria Kyriakidou)
Genome assembly is a hierarchical data structure that maps the sequence data to a putative
reconstruction of the target
Two types
oDe novo: the process of reconstructing the DNA sequence of an organism from its
sequence reads alone
No reference genome
Necessary for novel genome
Issues: length of the sequenced reads, errors, repeats
oReference based
Difficulties in assembling genomes
oBiological: high ploidy, heterozygosity, repetitiveness
oSequencing: large genomes, no perfect sequence yet
oComputational: large genomes, complex structures
oAccuracy: hard to assess correctness
Hierarchical structure of assembly: reads -> contigs -> scaffolds
oReads: fragments of original DNA with sequenced ends
oContigs: align reads to build contigs -> then align the cotigs to get a consensus contig
Major problem: repeats
oScaffolds: use of additional information to orient and connect contigs (paired end, mate
pair, restriction maps)
Paired end reads 100-500 bp insert
Mate pairs: 2-20 kb insert
Algorithms
oAll graph-based -> simplify assembly
Read layout
Overlap graph (overlap-layout consensus)
All versus all pairwise comparison of reads
Computationally very expensive
Does not scale well
Most fragmented assembly algorithms consist of the following steps
Overlap: finding potentially overlapping reads - alignment
(computationally intensive)
Layout: finding the order of the reads along DNA (graph
simplification)
Consensus: deriving the DNA sequence of reads along DNA
(sequence)
de Bruijn graph
Concept in combinatorial mathematics
Representation of sequence based on short words (k-mers)
Overlaps between words
Procedure
Split the reads into k-mer size chunks
K-mer is a short substring of reads (for this example, k=3)
Dk = (V,E)
find more resources at oneclass.com
find more resources at oneclass.com
Document Summary
Lecture portion: introduction to genome assembly (lecturer: maria kyriakidou) Genome assembly is a hierarchical data structure that maps the sequence data to a putative reconstruction of the target. Two types: de novo: the process of reconstructing the dna sequence of an organism from its sequence reads alone. Issues: length of the sequenced reads, errors, repeats o. Difficulties in assembling genomes o o o. Computational: large genomes, complex structures: accuracy: hard to assess correctness. Hierarchical structure of assembly: reads -> contigs -> scaffolds. Reads: fragments of original dna with sequenced ends. Contigs: align reads to build contigs -> then align the cotigs to get a consensus contig o o. Scaffolds: use of additional information to orient and connect contigs (paired end, mate pair, restriction maps) Algorithms: all graph-based -> simplify assembly. Most fragmented assembly algorithms consist of the following steps. Overlap: finding potentially overlapping reads - alignment (computationally intensive) Layout: finding the order of the reads along dna (graph simplification)